
Knowledge Library & FAQs
On this page you'll find general information and answers to common questions regarding conducting clinical research at the University of Calgary. If you have a question that isn't listed, please contact our office. Information is organized into the following categories:
- Case Report Forms & Source Documentation
- Clinical Trial Overhead Requirements
- Essential Documents
- Facilities & Equipment
- Health Canada Applications
- Investigational Product (IP)
- Monitoring
- Participant Recruitment, Eligibility & Enrolment, & Consent
- Project Closure and Archiving
- Protocol Adherence
- Quality Assurance for Clinical Trials
- Study Personnel, Responsibilities & Qualifications
Case Report Forms & Source Documentation
- There are many ways to document evidence of investigator oversight. A few examples include:
- Team meetings – include the agendas and documented attendance
- Eligibility checklists – have the investigator sign off on eligibility
- Printed out emails regarding any questions addressed to the investigator about the study or a participant’s care
- Serious Adverse Events – have the investigator sign off and comment on causality, severity, expectedness and whether it need to be reported.
- Abnormal lab results – have the investigator comment on whether it is clinically significant or not, any changes to care, etc.
- Review of CRA/Monitor visit letters and newsletters – have the investigator sign and date these communications.
Health Canada is looking for evidence to show that the investigator had significant involvement with the study.
GCP says that source data should be attributable, legible, contemporaneous, original, accurate, and complete (ALCOA-C). Changes to source data should be traceable, should not obscure the original entry, and should be explained if necessary (for example, via an audit trail).
If a correction needs to be made, the original record must still be legible: Make a single line through the error – never use correction fluid, multiple cross-outs or marker pen to obscure the original record. Record the correction next to the original record. Provide a brief comment why the change is required (as appropriate). Initial the change so that it is clear that the correction is deliberate. Record the date of the correction next to the initials so that there is a record of when the change was made.
The CRF should only contain identifiers approved by the REB to be collected. If any identifiable information is required, the researcher must be able to justify why they are unable to meet their research objectives without using the identifier. If this is the case, we recommend using the least identifiable form of that information. Examples can be found in section 2.2.2 of CIHR Best Practices for Protecting Privacy in Health Research.
An example could include Date of Birth. In a study with adults, you can probably get away with age or month and year of birth, but in a research study with newborns, you may need the full date and time of birth for eligibility determination.
Essential Documents
Essential documents can be in an electronic format. Electronic records should be maintained and retained in accordance with section C.05.012 of the Health Canada regulations. When creating an electronic essential documents binder, it is important that you have an SOP to ensure standard naming conventions, file systems, etc. It is recommended that it be placed inside a shared drive where only those with approval to access have access. Ensuring that the documents in the shared drive are the only source to avoid duplications. The system used must be backed up and recoverable, which is why it is best to be housed on an institutional drive and not your desktop or computer drive.
Although an electronic system could help save paper, you may still require some paper files. Examples include any documents requiring wet ink signatures.
Section 8 of Good Clinical Practice (GCP) lists all of the documents you will need as a site. The current version of GCP can be found here.
Section 8 of Good Clinical Practice (GCP) lists all of the documents you will need to collect from your sub-sites as the sponsor. The current version of GCP can be found here.
The easiest way to organize your documentation when you are the sponsor is to separate your site binder (UCalgary) from your sponsor binders.
If you have the funding for two coordinators, it will be easier to keep files organized if you have one site coordinator who takes care of running the study at your site and one multi-centre coordinator who coordinates the entire study and collects the documentation from all of the sites.
As a member of N2, the University has access to a set of fully vetted SOPs. They are available to all University research teams and can be found here.
If you feel that an SOP is not a reflection of your process, you may create an addendum the SOP. Here is a simple example of what your addendum could look like.
-
Current section of SOP008_08
Section 5.1.11 Sign and date the ICF as the person who conducted the informed consent discussion. Obtain any other signatures/dates, as indicated on the ICF.
-
Amendment to SOP008_08
Section 5.1.11 Due to the complexity and risks of the protocol, only a physician may conduct the informed consent discussion and sign and date the ICF.
Facilities and Equipment
All service, maintenance, calibration records, as well as the product manual for critical pieces of equipment used for the clinical trial should be collected and maintained throughout the study.
The manual calibration of certain pieces of equipment or instruments (e.g. body weight scales) does not generate a certificate or a print-out of the calibration data to demonstrate that the calibration was indeed performed and successful. In those circumstances, the QI should retain the calibration procedure and a log stating the following information:
- dates of calibration
- device details (type, supplier, and purchase date)
- person responsible for the instrument
- person who performed calibrations
Approved specifications for calibration should also be documented and a record of the actual calibration results kept.
For hospital owned equipment, a letter from AHS indicating their care and maintenance of the equipment is sufficient. A template letter can be found in N2 SOP025.
Health Canada Applications
Studies that require a CTA or ITA submission to Health Canada include:
-
Drug studies
- Phase I-III clinical trials
- Comparative bioavailability trials
- Clinical trials involving marketed drugs, where the proposed trial is outside of the parameters of the authorized NOC or DIN application, e.g., one or more of the following is different
- indication(s) and clinical use
- target patient population(s)
- route(s) of administration
- dosage regimen(s)
-
Device studies
- Unlicensed class II, III and IV medical devices imported and/or sold in Canada for research purposes.
- For help determining if your device requires an Investigational Testing Authorization application, refer to the ITA flow chart.
-
Natural Health Product studies
- If an NHP previously approved by Health Canada is being tested for a condition of use not captured on the product label.
- For NHPs that are not yet approved by Health Canada and for which additional efficacy and safety evidence is required before marketing can be authorized.
- For NHPs with no prior history of use in humans. For example, new isolates or new extracts on their own or in combination with other medicinal ingredients proven to be safe.
-
Note:
If the study includes a combination of the study types listed above, you will need to apply for approval from all branches involved.
According to Health Canada a “Qualified Investigator (QI)” must be a physician or dentist and a member in good standing of a professional medical or dental association as defined in Part C Division 5 of the Food and Drug Regulations.
- All UCalgary Investigator-Initiated clinical trials must list the sponsor as the Governors of the University of Calgary. The University does not allow for Investigator-Sponsors due to the risk to the Institution.
- Name the Governors of the University of Calgary as the Sponsor
- Name the Associate Vice President (Health Research) (currently Dr. Marcello Tonelli)as the Senior Executive Officer (box 93)
- Once completed, the application along with all of the documents should be forwarded to the Quality Assurance and Regulatory Compliance Specialist, Jenna Dobry to review for completeness.
- Once complete, the QA and Regulatory Compliance Specialist will forward on for AVP (Health Research)’s signature. The QI is responsible for collecting the other signatures.
- To ensure completeness, refer to the CTA checklist.
1. CTA-A must be filed when the proposed amendments to the protocol (taken from the HC Guidance Document):
- Affect the selection, assessment, or dismissal of a clinical trial subject;
- Affect the evaluation of the clinical efficacy of the drug;
- Alter the risk to the health of a clinical trial subject;
- Affect the safety evaluation of the drug; or
- Extend the duration of the treatment.
2. Notifications include the following (taken from the Health Canada Guidance Document):
- Changes to the protocol that do not affect the safety of the trial participants and which would not be considered an amendment under section 2.4. Examples include, but are not limited to the following:
- Minor changes to the inclusion and exclusion criteria, such as laboratory chemistry cut-off values that reflect clinical practice and improve the safety of clinical trial subjects;
- Increasing the screening period or other administrative changes to accommodate logistical constraints in study conduct that do not affect the safety of the trial participants;
- Changes to administrative information such as new contact names and numbers and addresses of individuals, organizations, or other entities, involved in the conduct of the trial;
- Updating the ICF with new safety information that does not require a protocol amendment;
- Annual Investigator Brochure updates (see section 2.8.5).
- Changes to Quality (Chemistry and Manufacturing) information that does not affect the quality or safety of the drug (refer to section 2.4.2 CTA-As and CTA-Ns: Quality (Chemistry and Manufacturing)).
- Changes as described in sections 2.7.1.1 (Refusals); 2.7.5. (Importation of Clinical Trial Drugs); and, 2.8.1 (Premature Discontinuance of a Trial).
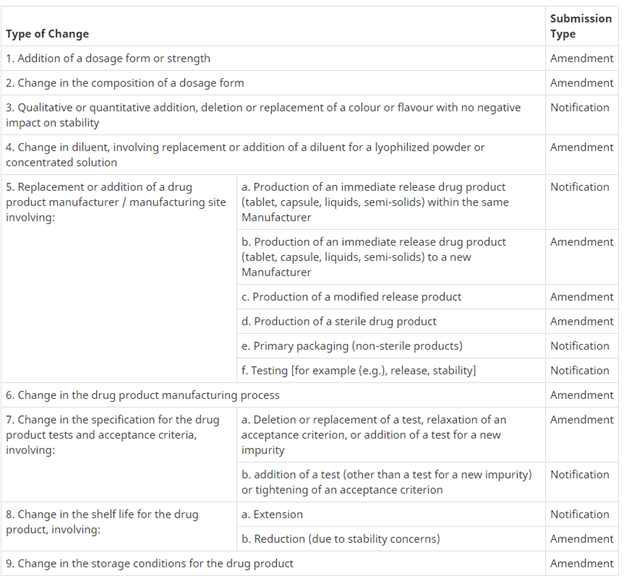
The table below provides more information about whether your revision is an amendment or a notification.
{kind=link}
Investigational Product
- The following records will be requested during an inspection:
- Temperature logs and any deviations
- IP labels
- Calibration records for all of the equipment
- Shipping receipts
- Confirmation of receipt and/or temperature log data to the sponsor is filed. Any deviations / defects upon receipt were documented
- Destruction documentation
- Accountability logs (IP lot #, dose, amount, expiration dates, dispensing date, any calculations, who dispensed to, any returns, etc.)
- Unblinding procedures
- IP handling manual
- Certificate of analysis for the IP
- Randomization records
- Communication
- Health Canada Part C Division 5 indicates that the drug bears a label that sets out the following information in both official languages:
- a statement indicating that the drug is an investigational drug to be used only by a qualified investigator;
- the name, number or identifying mark of the drug;
- the expiration date of the drug;
- the recommended storage conditions for the drug;
- the lot number of the drug;
- the name and address of the sponsor;
- the protocol code or identification; and
- if the drug is a radiopharmaceutical as defined in section C.03.201, the information required by subparagraph C.03.202(1)(b)(vi).
For safety reasons it is not recommended to store IP in your offices. There have been reports where drug was stolen from an office.
Any IP that is a controlled substance can only be stored in the research pharmacy where they have a license to hold them.
If IP is stored in your office, it must be stored and monitored under secure conditions (lock and key), only available to people delegated the role to store and dispense IP. The IP must be stored as indicated in the Investigator’s Brochure (IB) (fridge, freezer, ambient, or warm/hot). You must have a plan to deal with any temperature excursions, proper destruction, return to suppliers if required, etc. For these reasons, we highly recommend the use of the research pharmacy who is used to dealing with these issues.
Yes, if you are conducting a phase IV drug study, you are still required to follow Health Canada Part C Division 5 regulations. It is also important to know that your study could be chosen for inspection by Health Canada.
Monitoring
- The Clinical Trials Office under CCCR offers monitoring services on a fee for service basis. See the Monitoring Services page for more details
The risk of the study determines the type of monitoring required. A risk assessment is included as part of our fee for service monitoring or you can refer to the Risk-Based Monitoring templates on the Quality Assurance : here.
Participant Recruitment, Eligibility & Enrolment, & Consent
Confirming eligibility is considered a medical decision by Health Canada. Although the Coordinator may complete an eligibility checklist by reviewing medical records, etc., a physician investigator must make the final confirmation of eligibility.
The easiest way to document eligibility is to create an eligibility checklist that has a spot for the investigator to sign off confirming that the patient is eligible to participate in the study.
Each eligibility criterion must have a source document to back it up. If there is no documented proof (medical records, questionnaires, documentation of any discussions with the participant about medical history, lab results, etc.) then the criterion cannot be confirmed.
If a criterion requires using an app to complete a calculation, we recommend you take a screen shot and place a copy in the participant study records.
Section 4.8 of GCP describes the informed consent of trial participants. It indicates that the investigator or delegate should discuss the trial with the participant, allow the participant time to ask questions and have them answered to their satisfaction, provide ample time for the participant to discuss and decide if they wish to participate, and ensure that participants are given a copy of the signed and dated consent form prior to participation in the study.
The easiest way to ensure you have documented the informed consent process appropriately is through the use of a checklist that is completed at the time of consent and signed and dated by the person obtaining consent. A copy of a consent process template can be found here.
The research team should ensure that participants are informed of any new information that becomes available throughout the study.
Discussions about the study and the participant’s consent to continue in the study should be documented in the participant’s study records.
Ongoing consent is especially important when a participant has a disease or illness where their capacity to give consent may come and go.
Whenever a change is made to a consent form that may impact participants’ consent to continue in the research project, they should be asked to re-consent. A few examples include: updated safety information, additional study visits, change in treatment arms, etc. The REB may request changes to the consent form at any time and request participants to be re-consented.
When the REB approves the use of verbal consent, you must document the consent process. This can be done by adding notes about the consent discussion, the questions asked, and the actual verbal consent to the consent check list. If there is no risk to the participant, they may be given written documentation about what their involvement includes.
If consent is obtained verbally over the phone, it would be appropriate to have a witness on the call, or, in some cases, a recording of the consent process. A recording should not be made if it may compromise the participant’s safety or confidentiality. An example would be a study recruiting participants conducting illegal activities.
Do I need to keep the consent form?
Yes, although participants are able to request their data be removed from a study when they withdraw, all consenting documentation needs to be kept. The data is removed from the analysis but the consent stored for safety purposes. For example, if there is a recall on the IP and you need to call the participant in for follow up testing.
The person who conducted the informed consent discussion with the participant signs the consent form. Typically this is a responsibility delegated to the Coordinator, but if the Investigator obtained consent, they would sign.
A witness is used when the participant (or the substitute decision maker) is unable to read. In this case an impartial witness who is not affiliated with the study on the study delegation log should be present. The witness is attesting that the information in the consent form was accurately explained to the participant, that the participant (substitute decision maker) apparently understood, and that consent was given.
According to TCPS2, the REB may allow research that involves medical emergencies to be carried out without the consent of participants, or of their authorized third party, if all of the following apply:
- A serious threat to the prospective participant requires immediate intervention.
- Either no standard efficacious care exists, or the research offers a realistic possibility of direct benefit to the participant in comparison with standard care.
- Either the risk is not greater than that involved in standard efficacious care, or it is clearly justified by the prospect for direct benefits to the participant.
- The prospective participant is unconscious or lacks capacity to understand the risks, methods and purposes of the research project.
- Third party authorization cannot be secured in sufficient time, despite diligent and documented efforts to do so.
- No relevant prior directive by the participant is known to exist.
-
Examples are: (Note: It is the REB’s decision if deferred consent is approved or not)
- Paramedic research where the treatment has been randomized by city/zone and all patients picked up with the condition are recruited into the study automatically.
- A time sensitive stroke study where there is no substitute decision maker available and the patient is unconscious or unable to provide informed consent.
-
For studies with US involvement (FDA, NIH, HHS, etc.), public consultation is required.
The “Exception from Informed Consent requirements for Emergency Research” guidance document, advises that consultation is required (including, where appropriate, consultation carried out by the IRB) with representatives of the communities in which the clinical investigation will be conducted and from which the subjects will be drawn. The guidance document can be found here.
-
NOTE:
When a participant or substitute decision maker is available to provide deferred consent, you must provide a debriefing of why you enrolled the participants without obtaining consent and answer any questions or concerns the participant/substitute decision maker may have.
Once a participant regains capacity to consent, they should be asked to provide their consent to continue participating in the research study.
If the participant chooses not to consent, they are withdrawn from the study and, like you would with any withdrawal, you should discuss their level of withdrawal. For example, can you use the data collected up to that point or should it be withdrawn.
Project Closure and Archiving
The standard Project closure and archiving process begins when the Sponsor confirms with the research team that all work on the clinical trial has been completed, and the relationship between the researcher and the Sponsor on the project has ended. The CCCR provides study closure and archiving services to both industry funded and non-industry funded projects, including guidance regarding:
- Financial close-out
- Allocation of residual funds
- Project closure coordination with ethics and legal teams
For detailed information regarding the archiving and project closure processes, policies and requirements, click here.
Protocol Adherence
Health Canada will review your protocol to ensure it contains all of the elements requested in GCP. Section 6 of GCP lists all of the GCP requirements for a clinical trial protocol.
Another option is to use a protocol checklist like the SPIRIT checklist (If you use this checklist, remember to give appropriate credit).
No, you cannot implement an amendment before receiving REB/Health Canada approvals.
The only exception is if the amendment is to remove an immediate danger to the study participant(s). The investigator/sponsor must submit the amendment report within 15 days after implementation of the amendment.
Each study should have a protocol deviation log to track all deviations. The log should include the following information: date, participant ID, description of deviation, type of deviation (examples include ICF, inclusion/exclusion, study procedures, randomization, etc.), any additional comments and a signature line for the person tracking them.
The reason for tracking deviations are to identify any trends that may require an amendment or additional training.
My research includes participant interviews. The participant has said something interesting that I want to follow up on. Do I need to follow the script or can i ask additional questions?
You should always follow the REB approved interview script. That said, if something interesting is said that you wish the participant to elaborate on, you can follow up with a question not on the script. If however, you find yourself asking the same additional question(s) to most of the participants, you would be required to submit an amendment to the script for approval by the REB.
Study Personnel, Responsibilities & Qualifications
You can find a template here. The template should be modified to reflect the specific duties required for your study.
For example, if you are not collecting biological samples, you should remove it from the list of duties. Or if you are conducting MRIs, this should be added. There shouldn’t be any duties in the list that are not part of the procedures, and you should add any missing procedures.
- Any member of the study team with a major role in the study should be listed on the delegation of duties log. Examples include (but are not limited to):
- Investigators (incl. QI, Co-Is, and student investigators)
- Coordinators
- Research Assistants
- Research Pharmacist (drug studies)
- Lab Manager (if local laboratory tests are required)
- Research Nurses
The CHREB will look for completion of the Tri-Council Policy Statement, Part 2 (TCPS2 Core tutorial)
Alberta Health Sciences (AHS) will not release patient data without proof of privacy training. (AHS Information Privacy & IT Security Awareness)
NOTE: Service providers do not need to be included in the training requirements. By service provider, we are referring to individuals like a diagnostic imaging technician conducting study related scans, a respiratory technologist conducting PFTs, lab techs drawing blood for study visits, etc. Service providers are not part of the study team, they are only providing a service to the study team. Other areas not requiring training could be data entry clerks, administrative assistants printing off documents for study visits, clinic clerks registering participants into the clinic, etc.
CVs should be on file for all members of the study team named on the delegation of duties log.
You will require the licenses of all professionals involved in the study. Examples include any MDs, Nurses, licensed technicians, pharmacists, etc.
NOTE: CVs should be signed and dated and updated with any new training, roles, etc. every 2 years. If the study is a multi-year project, you should collect and keep the licenses for each year the person is involved in the study.
All medical care and decisions should only be delegated to physicians on the study team. This includes, but is not limited to, confirmation of eligibility, assessment of SAEs, physical exams, etc
There are many ways to document training:
- Training certificates (CITI Canada or TCPS2 certificates are an example)
- Self-guided training – reading the material yourself and documenting that you have read and understood and signed and date the document (SOP training would be an example)
- In person training – print a copy of the agenda and slides (if any). Ensure you sign the attendance form (CCCR lunch and learns are an example)
- Team meeting training – add the training to the meeting agenda and take attendance (protocol amendment training is an example)
Above and beyond the CVs, professional licenses and training records, any study specific training should be on file. Examples include, but not limited to: eCRF training, phlebotomy training (if not conducted by a nurse or lab technician), lab manual training for any members involved with biological samples. Consent process training if this is the first time they are consenting participants, etc.